Phân tích mã gen SARS-CoV-2 bằng một số thư viện Python
Bài đăng này đã không được cập nhật trong 5 năm
Đại dịch COVID-19 là một đại dịch bệnh truyền nhiễm với tác nhân là virus SARS-CoV-2 mới xuất hiện gây ra hội chứng hô hấp cấp tính nghiêm trọng cho sức khỏe cộng đồng toàn cầu ảnh hưởng nặng nề đến các nền kinh tế kể từ khi nó được xác định vào cuối tháng 12 năm 2019 tại Trung Quốc. Bằng cách sử dụng một số thư viện của Python, chúng ta có thể trích rút ra được một số hiểu biết chung từ dữ liệu gen của loại virus này.
Tìm hiểu dữ liệu bộ gen
Trước khi đọc tiếp, trong trường hợp mọi người quên khai niệm của DNA và RNA thì có thể đọc tại đây Do You Know sự khác nhau giữa DNA và RNA?
Mã di truyền và DNA codon
Mã di truyền là tập hợp các quy tắc được tế bào sống sử dụng để dịch thông tin được mã hóa bên trong vật liệu di truyền (trình tự DNA hoặc mRNA của bộ ba nucleotide hay còn được gọi là codon) thành protein.
Mã di truyền tiêu chuẩn thường được biểu diễn dưới dạng bảng codon RNA bởi vì, khi protein được tạo ra trong tế bào bởi ribosome, là mRNA sẽ chỉ đạo tổng hợp protein. Trình tự mRNA được xác định bởi trình tự của DNA bộ gen.
Một số tính năng của codon có thể liệt kê như sau:
- Hầu hết các codon đều chỉ định một loại axit amin
- Bộ ba stop codon đánh dấu sự kết thúc của một protein
- Một start codon, AUG, đánh dấu sự bắt đầu của protein và cũng mã hóa axit amin methionine
Tài liệu tham khảo: https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
Phân tích mã gen SARS-CoV-2 bằng thư viện Biopython
So sánh và đối chiếu các loại vi rút gây ra COVID-19, SARS và MERS và tìm hiểu sự khác biệt và tương đồng chính giữa các loại virus có liên quan. Đây là một cuộc thi trên Kaggle có tên là Coronavirus Genome Sequence, để tìm hiểu thêm về chủ đề cũng như một số cách tiếp cận khác mọi người có thể truy cập tại https://www.kaggle.com/paultimothymooney/coronavirus-genome-sequence.
Chuẩn bị dữ liệu và cài đặt các thư viện
Dữ liệu được sử dụng trong bài được cung cấp ở cuộc thi trên, mọi người có thể tải về tại đây https://www.kaggle.com/paultimothymooney/coronavirus-genome-sequence#MN908947.fna. Dữ liệu vừa được đề cập là giải trình tự RNA di truyền của một mẫu dịch rửa phế quản phế nang từ bệnh nhân đã xác định được một dòng vi rút RNA mới từ họ Coronaviridae, được chỉ định ở đây là coronavirus ‘WH-Human 1’ (và còn được gọi là ‘2019-nCoV’).
Tiếp theo, để thực hiện ví dụ trên, chúng ta cần đến thư viện Biopython cũng như một số các thư viện phụ trợ như numpy, pandas, matplotlib, os cũng như seaborn mà có thể dễ dàng cài đặt bằng cách sử dụng pip
Sau khi cài đặt, chúng ta import thư viện là đọc dữ liệu bằng đoạn code sau:
import numpy as np
import pandas as pd
pd.plotting.register_matplotlib_converters()
import matplotlib.pyplot as plt
%matplotlib inline
import seaborn as sns
import os
#Load data
from Bio import SeqIO
for sequence in SeqIO.parse('data/covid19-MN908947.fna', 'fasta'):
print(sequence.seq)
print(len(sequence), 'nucliotides')
Bên cạnh đó chúng ta có thể đọc dữ liệu có thể điều chỉnh (align) được bằng cách như sau:
from Bio.SeqRecord import SeqRecord
from Bio import SeqIO
DNAsequence = SeqIO.read('data/covid19-MN908947.fna', 'fasta')
DNAsequence
Chuyển đổi dữ liệu
Vì trình tự đầu vào là FASTA (DNA) và SARS-CoV-2 là loại vi rút RNA, bởi vậy nên chúng ta cần:
- Phiên mã DNA thành RNA (ví dụ như ATTAAAGGTT sẽ được chuyển thành AUUAAAGGU)
- Dịch RNA thành chuỗi acid amin (ví dụ như AUUAAAGGUU sẽ được chuyển thành IKGLYLPR * Q) Hai quá trình trên sẽ được trình bày dưới đây:
Chuyển đổi DNA thành RNA
Để chuyển đổi, chúng ta sử dụng đoạn mã như sau:
DNA = DNAsequence.seq
mRNA = DNA.transcribe()
print(mRNA)
print(f'Size: {len(mRNA)}')
Phần đầu của dữ liệu thu được sẽ như thế này:
AUUAAAGGUUUAUACCUUCCCAGGUAACAAACCAACCAACUUUCGAUCUCUUGUAGAUCUGUUCUCUAAACGAACUUUAAAAUCUGUGUGGCUGUCACUCGGCUGCAUGCUUAGUGCACUCACGCAGUAUAAUUAAUAACUAAUUACUGUCGUUGACAGGACACGAGUAACUCGUCUAUCUUCUGCAGGCUGCUUACGGUUUCGUCCGUGUUGCAGCCGAUCAUCAGCACAUCUAGGUUUCGUCCGGGUGUGACCGAAAGGUAAGAUGGAGAGCCUUGUCCCUGGUUUCAACGAGAAAACACACGUCCAACUCAGUUUGCCUGUUUUACAGGUUCGCGACGUGC ......
Chuyển đổi RNA thành chuỗi các amino acid
Protein được tạo thành từ các đơn vị nhỏ hơn gọi là acid amin, chúng được gắn vào nhau trong một chuỗi dài. Có 20 loại axit amin khác nhau có thể kết hợp để tạo ra protein. Trình tự các axit amin quyết định cấu trúc 3D độc đáo của mỗi protein và chức năng cụ thể của nó.
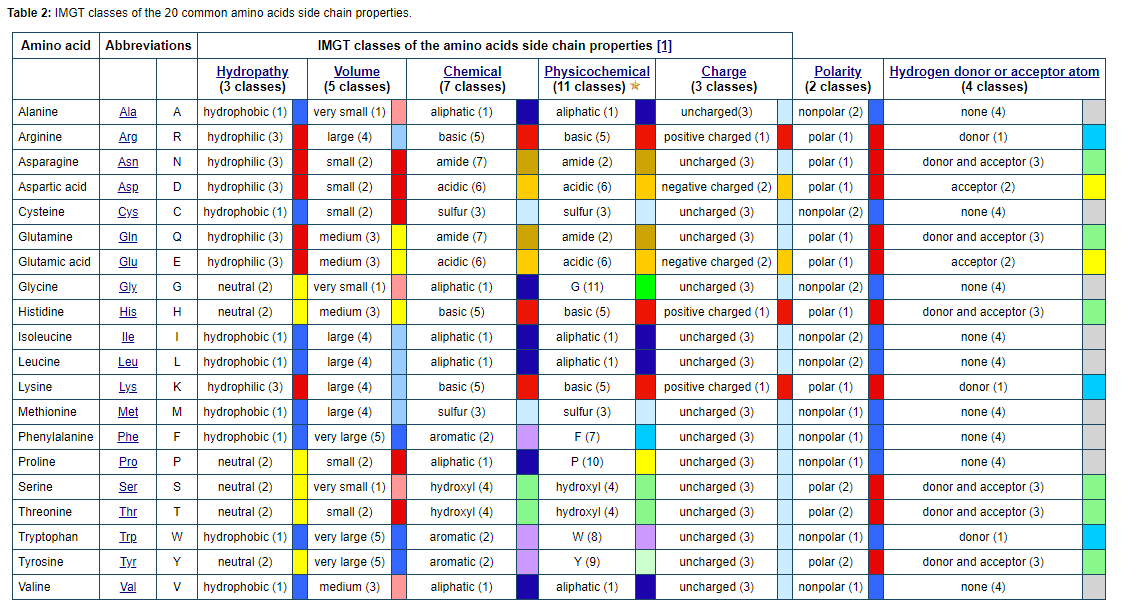
Hình ảnh trên lấy từ trang http://www.imgt.org/IMGTeducation/Aide-memoire/_UK/aminoacids/IMGTclasses.html
Để tìm hiểu về 20 loại amino acid trên mọi người có thể xem tại https://www.hornetjuice.com/amino-acids-types/
Để chuyển đổi RNA thành chuỗi các amino acid chúng ta thực hiện như sau:
# Load the condo table
from Bio.Data import CodonTable
print(CodonTable.unambiguous_rna_by_name['Standard'])
# Transform RNA sequence to amino acid sequence
aacid = mRNA.translate(table=1, cds=False)
print('Amino Acid', aacid)
print("Length of Protein:",len(aacid))
print("Length of Original mRNA:",len(mRNA))
#3. Remove unfunctional protein
# short-length proteins are likely to play a little biological role --> removed
# 20 amino acids long is the smallest known functional protein
Loại bỏ một số protein không chức năng
Một số protein được gọi là không chức năng (non-functional protein) bởi chúng đóng góp một phần không đáng kể đến việc quyết định tính trạng của virus vậy nên chúng ta có thể bỏ qua chúng khi sử dụng dữ liệu các amino acid trên.
Một số tiêu chuẩn để xác định một protein có phải không chức năng bằng cách xem xét rằng protein dài ngắn có thể đóng một vai trò sinh học nhỏ nên là protein không chức năng. Bởi vậy các acid amin có độ dài 20 là protein chức năng nhỏ nhất được biết đến sẽ bị loại bỏ.
Để thực hiện điều đó, chúng ta thực hiện như sau:
#Identify all the Proteins (chains of amino acids)
proteins = aacid.split('*')
print(f"Before removing, number of proteins: {len(proteins)}")
proteins = [p for p in proteins if len(p) >=20]
print(f"After removing, number of proteins: {len(proteins)}")
Trích xuất đặc trưng từ protein bằng Protparam
ProtParam cung cấp một số công cụ có sẵn có thể kể đến như sau:
count_amino_acids: đếm axit amin tiêu chuẩn, trả về một dict.get_amino_acids_percent: Tính hàm lượng acid amin theo phần trăm.molecular_weight: Tính MW (trọng lượng phân tử) từ trình tự Protein.aromaticity: Tính giá trị độ thơm của protein theo Lobry & Gautier (1994, Nucleic Acids Res., 22, 3174-3180).flexibility: Thực hiện phương pháp linh hoạt của Vihinen và các cộng sự *(1994, Protein, 19, 141-149). *isoelectric_point: Tính điểm đẳng điện (Isoelectric point, pI) của protein.secondary_structure_fraction: Phương thức này trả về một danh sách các phần axit amin có xu hướng ở dạng xoắn, dạng turn hoặc dạng sheet.- Các axit amin ở dạng xoắn: V, I, Y, F, W, L.
- Các axit amin ở dạng turn: N, P, G, S.
- Các axit amin ở dạng sheet: E, M, A, L.
Để tính toán các giá trị trên chúng ta thực hiện như sau:
poi_list = []
MW_list = []
from Bio.SeqUtils import ProtParam
for record in proteins:
print("\n")
X = ProtParam.ProteinAnalysis(str(record))
POI = X.count_amino_acids()
poi_list.append(POI)
MW = X.molecular_weight()
MW_list.append(MW)
print("Protein of Interest = ", POI)
print("Amino acids percent = ",str(X.get_amino_acids_percent()))
print("Molecular weight = ", MW_list)
print("Aromaticity = ", X.aromaticity())
print("Flexibility = ", X.flexibility())
print("Isoelectric point = ", X.isoelectric_point())
print("Secondary structure fraction = ", X.secondary_structure_fraction())
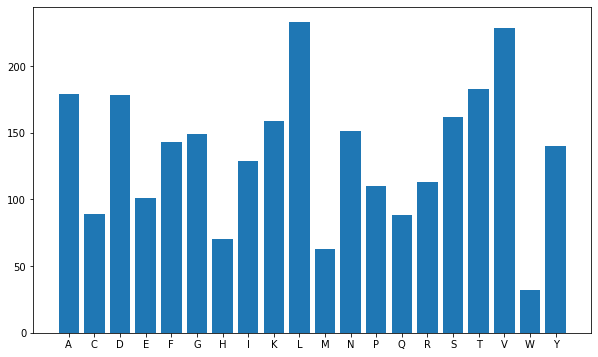
Để trực quan hóa ta sự dụng đoạn mã sau:
MoW = pd.DataFrame(data = MW_list,columns = ["Molecular Weights"] )
#plot POI
poi_list = poi_list[48]
plt.figure(figsize=(10,6));
plt.bar(poi_list.keys(), list(poi_list.values()), align='center')
Kết quả thu được có thể trông như hình sau:
So sánh RNA của Coronavirus ở người
Để bắt đầu thực hiện, việc đầu tiên cần làm là đọc dữ liệu:
sars = SeqIO.read("data/sars.fasta", "fasta")
mers = SeqIO.read("data/mers.fasta", "fasta")
cov2 = SeqIO.read("data/cov2.fasta", "fasta")
print(len(sars.seq), len(mers.seq), len(cov2.seq))
Để có thể quan sát dễ hơn dữ liệu vừa đọc chúng ta có thể sử dụng đã được giới thiệu ở bài trước như sau:
!squiggle data/cov2.fasta data/sars.fasta data/mers.fasta --method=gates --separate
Căn chỉnh trình tự (Sequence alignment)
Quá trình căn chỉnh trình tự (Sequence alignment) sắp xếp hai hoặc nhiều trình tự (của trình tự DNA, RNA hoặc protein) theo một trật tự cụ thể để xác định vùng tương đồng giữa chúng. Từ đó, chúng ta có thể suy ra nhiều thông tin như những đặc điểm nào được bảo tồn giữa các loài, các loài khác nhau gần gũi về mặt di truyền như thế nào, các loài tiến hóa như thế nào, v.v.
Vậy nên căn chỉnh trình tự theo cặp chỉ so sánh hai trình tự tại một thời điểm và cung cấp sự liên kết trình tự tốt nhất có thể.
Để thực hiện chúng ta sử dụng đoạn mã sau:
# Alignments using pairwise2 alghoritm
sars_cov = pairwise2.align.globalxx(sars.seq, cov2.seq, one_alignment_only=True, score_only=True)
print('SARS/COV Similarity (%):', sars_cov / len(sars.seq) * 100)
mers_cov = pairwise2.align.globalxx(mers.seq, cov2.seq, one_alignment_only=True, score_only=True)
print('MERS/COV Similarity (%):', mers_cov / len(mers.seq) * 100)
mers_sars = pairwise2.align.globalxx(mers.seq, sars.seq, one_alignment_only=True, score_only=True)
print('MERS/SARS Similarity (%):', mers_sars / len(sars.seq) * 100)
Có thể align không có sẵn trong pairwise2 và sẽ gây lỗi, vậy nên để kiểm tra chúng ta có thể sử dụng đoạn mã sau trước khi chạy đoạn mã trên.
from Bio import pairwise2
# check if pairwise2.align is exist
pairwise2.align??
Kết Luận
Ví dụ trên đã trình bày một số kiến thức cơ bản về DNA, RNA và acid amin cũng như cách nó triển khai các thông tin di truyền thành tính trạng và bên cạnh đó trình bày một ví dụ nhỏ về việc so sánh và đối chiếu các loại vi rút gây ra COVID-19, SARS và MERS và tìm hiểu sự khác biệt và tương đồng chính giữa các loại virus có liên quan.
Các nhà khoa học tại Đại học Liverpool, những người cũng đã tham gia Liên minh giải trình tự gen toàn bộ đã giải thích rằng việc hiểu mã di truyền được lấy từ các mẫu cá nhân của bệnh nhân bị nhiễm bệnh sẽ cho phép họ hiểu:
- Ai đang bị bệnh
- Loại bệnh mà họ mắc phải, và tại sao
- SARS-CoV-2 gây bệnh như thế nào
- Hệ thống miễn dịch của bệnh nhân có phản ứng quá mức hay không
- Liệu các chủng khác nhau của virus đó có đang bắt đầu xuất hiện hay không.
Vậy nên, việc tìm hiểu mã di truyền, bao gồm đối sánh dữ liệu giải trình tự RNA và rất nhiều quá trình phức tạp hơn đóng vai trò rất quan trọng trong việc đẩy lùi dịch bệnh. Bài viết đến đây kết thúc, cảm ơn mọi người đã giành thời gian đọc.
Tài liệu tham khảo
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7139247/
- https://www.greelane.com/vi/khoa-học-công-nghệ-toán/khoa-học/dna-versus-rna-608191/
- https://www.kaggle.com/paultimothymooney/coronavirus-genome-sequence
- https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
- https://www.hornetjuice.com/amino-acids-types/
- https://biopython.org/docs/1.75/api/index.html
All rights reserved
