Xác định các mẫu động vật khảo cổ tự động từ quang phổ MALDI-ToF (Phần 1)
Bài đăng này đã không được cập nhật trong 5 năm
1. Lời mở đầu
Bài viết được tham khảo và dịch từ bài báo khoa học How to get your goat: automated identification of species from MALDI-ToF spectra.
Việc phân loại các mẫu động vật khảo cổ thường đạt được thông qua việc kiểm tra thủ công quang phổ trong thời gian bay / thời gian ion hóa của tia laser hỗ trợ bằng quang phổ (MALDI-ToF). Đây là một quá trình tốn nhiều thời gian, đòi hỏi năng lực chuyên môn cao và không tạo ra thước đo độ tin cậy trong việc phân loại. Bài báo giới thiệu một phương pháp mới, tự động phân loại mẫu MALDI-ToF, miễn là có sẵn các chuỗi collagen cho từng loài ứng viên cần phân loại. Phương pháp tiếp cận lấy một tập hợp các khối lượng peptit từ dữ liệu chuỗi để so sánh với dữ liệu mẫu, được thực hiện bằng tương quan chéo. Ở bài viết này, chúng ta sẽ đi tìm hiểu MALDI-ToF là gì và nó được ứng dụng trong bài toán phân loại này ra sao nhé!
2. MALDI-ToF là gì?
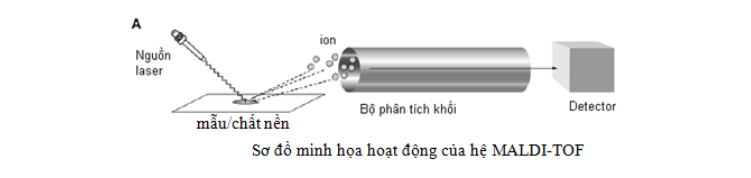
Đầu tiên, mình sẽ giải thích qua 1 chút về phương pháp khối phổ (MS - Mass spectrometry) là một kỹ thuật phân tích hóa học giúp xác định hàm lượng và loại chất hóa học có trong một mẫu vật bằng cách đo tỷ lệ khối lượng trên điện tích và số lượng của các ion pha khí. MALDI là thuật ngữ chỉ phương pháp ion hóa mẫu hấp thụ dựa trên sự hỗ trợ của các chất nền và năng lượng laser (Matrix-assisted laser desorption ionization, MALDI). Nguồn MALDI sử dụng các chất nền hỗ trợ để ion hóa mẫu dưới tác dụng của năng lượng laser lớn. Dòng Laser - ToF của hệ thống MALDI cung cấp bới SAI là dải sản phẩm ứng dụng mạnh mẽ và lý tưởng cho việc phân tích các phân tử hữu cơ phân đoạn lớn như phân tử sinh học và polyme.
Phương pháp MALDI-TOF và MALDI-TOF MS thường được sử dụng cho các mẫu protein đơn giản, và khá tinh sạch. Ưu điểm là có thể giải chuỗi axit amin mảnh peptit và đo chính xác khối lượng, nhưng nó lại không thích hợp cho việc phân tích hỗn hợp protein phức tạp.
3. Giới thiệu nghiên cứu
Phép đo khối phổ thời gian bay / thời gian ion hóa bằng tia laser hỗ trợ quang phổ (MALDI-ToF) (MS) là một công cụ được thiết lập để phân biệt giữa các loài từ các mảnh xương khi không có dấu hiệu hình thái (Buckley và các cộng sự, 2009), có nhiều ứng dụng trong an toàn thực phẩm (Di Francesco et al., 2018), pháp y (Francese, 2019) và khảo cổ học (Kuckova et al., 2009).
Các mẫu đồ tạo tác dựa trên collagen được phân tích theo cách này tạo thành MS với các đỉnh ‘đánh dấu’ đặc biệt tạo thành dữ liệu cơ sở của quá trình nhận dạng mẫu vật, vì các peptit collagen tạo thành ‘mã vạch’ của các đỉnh trong MS. Mặc dù nguyên tắc chỉ định các đỉnh cho collagen peptide và đưa ra kết quả là loài đơn giản, nhưng một số vấn đề cần phải được khắc phục để đi đến một nhận dạng thuyết phục. Các sửa đổi sau dịch mã của chuỗi collagen peptide có thể thay đổi vị trí mong đợi của các đỉnh trong phổ do sự thay đổi về khối lượng mẫu vật.
Phương pháp chuẩn bị mẫu dường như có ảnh hưởng đến việc các peptit 'bay' trong MALDI-ToF - một tỷ lệ các đỉnh được quan sát thấy nhỏ hơn nhiều so với dự kiến nếu tất cả các peptit được tạo ra bằng cách cắt collagen với Porcine trypsin (Quá trình này thường được gọi là phân giải protein bởi trypsin hoặc trypsin hóa). Thiết bị MALDI-ToF yêu cầu mức độ hiệu chuẩn, nghĩa là khối lượng quan sát được của đỉnh có thể là giá trị gần đúng lý thuyết.
Vì những lý do này, việc sử dụng trực tiếp dữ liệu chuỗi để xác định loài bằng MS cho đến nay là không thể thực hiện được, và cần có mức độ đánh giá chuyên môn và giải thích bằng chứng khác liên quan đến MS để đi đến nhận dạng mẫu loài. Ngoài ra, việc nhận dạng kết quả chỉ là định tính, do đó mức độ tin cậy trong việc nhận dạng và giá trị của các phân loại thay thế có thể có không thể được nêu ra bằng số.
Ở đây, bài báo đã mô tả một cách tiếp cận dựa trên phần mềm mới để xác định các loài từ MS. Với những tiến bộ trong phân tích protein và dữ liệu chuỗi ngày càng sẵn có, có thể dự đoán vị trí của các đỉnh trong MS dựa trên thành phần của các peptit trong một mẫu và sau đó so sánh các đỉnh quan sát được với vị trí đỉnh dự đoán. Sự phân bố các đồng vị của các peptit cũng có thể được dự đoán, cùng với các tác động lên khối lượng của bất kỳ biến đổi sau dịch mã nào. Khi đã có khối lượng dự đoán, các đỉnh trong MS có thể được căn chỉnh theo phân phối dự đoán bằng cách sử dụng kỹ thuật tương quan chéo đã được thiết lập. Điều này cho phép một đỉnh được chấp nhận là do peptit đang được xem xét, hoặc bị từ chối và loại bỏ khỏi phân tích tiếp theo.
Phần mềm đề cập tới được phát triển bằng ngôn ngữ lập trình mã nguồn mở R script và có sẵn để tải xuống dưới dạng pakage từ https://github.com/bioarch-sjh/bacollite. Bài viết lần sau, chúng ta sẽ cùng cài đặt và sử dụng công cụ này nhé =))
Phương pháp mà bài báo trình bày ở đây chủ yếu nhằm vào các mẫu khảo cổ dựa trên collagen như da, xương và tóc, nhưng nó có thể được áp dụng cho bất kỳ vấn đề phân loại MS nào có chuỗi protein cụ thể. Để chứng minh tính hiệu quả của phương pháp này, bài báo so sánh phân loại tự động với phân loại từ chuyên gia cho một nhiệm vụ điển hình mà MS sẽ được sử dụng. Bộ dữ liệu được đề cập bao gồm các mẫu giấy da không bị phá hủy từ mẩu cục tẩy (Fiddyment và cộng sự, 2015) của mỗi danh mục của "Lucas glo. cum A a 12th-century manuscript from St Augustine’s Abbey Canterbury". Bằng chứng lịch sử chỉ ra rằng những tấm giấy da này được làm từ da động vật chỉ từ ba loài: Cừu(Ovis), Bò(Bos), và Dê(Capra). Bài báo này trình bày so sánh cách phân loại tự động và phân loại của chuyên gia để minh họa tính hiệu quả của kỹ thuật mới.
4. Cách tiếp cận
Cách tiếp cận tự động này có 3 giai đoạn:
- Đầu tiên, sử dụng dữ liệu proteomic để tạo ra một tập hợp các khối lượng đánh dấu peptit 'lý thuyết' cho mỗi loài đang được xem xét. Sự phân bố khối lượng của các ion đối với mỗi điểm đánh dấu peptit được tính toán, có tính đến khả năng sửa đổi sau dịch mã và kết hợp các đồng vị của mỗi nguyên tố trong peptit.
- Thứ hai, sự hiện diện và độ lớn của các đỉnh ở khối lượng của mỗi peptit lý thuyết được đo bằng cách áp dụng tương quan chéo.
- Cuối cùng, dữ liệu tương quan cho tập hợp các điểm đánh dấu cho mỗi đơn vị phân loại ứng viên (ở đây là các loài) được kết hợp để đưa ra điểm số cho việc xác định, với việc phân loại dựa trên đơn vị phân loại có điểm cao nhất.
4.1 Tạo các đỉnh lý thuyết
Để thực hiện phân tích, phải có sẵn các chuỗi collagen của loài ứng viên. Đối với mỗi loài ứng viên, ba bước sau được thực hiện:
-
Phổ lý thuyết: Peptide trong MALDI-ToF được tạo ra bằng cách tiêu hóa collagen với Porcine trypsin, cắt giảm arginine (R) và lysine (K). Do đó, dễ dàng dự đoán các peptit sẽ được tạo ra bằng cách cắt bằng Porcine trypsin bằng cách đơn giản tách chuỗi collagen sau mỗi trường hợp của R và K trong chuỗi. Điều này tạo ra một tập hợp các peptit ứng viên có thể tính được khối lượng.
-
Sự phân cắt bị bỏ lỡ: Porcine trypsin không phải lúc nào cũng cắt ở mọi trường hợp R và K trong phân tử collagen, dẫn đến các peptit chứa các phân cắt bị bỏ sót. Các đánh giá ban đầu giả định rằng trypsin cắt giảm ở tất cả các trường hợp của các axit amin này và sự cắt giảm xảy ra chính xác giữa K hoặc R và axit amin tiếp theo. Khi chúng ta đã căn chỉnh các đỉnh MALDI-ToF quan sát được với tập hợp các peptit được phân cắt hoàn hảo, sau đó chúng ta có thể kiểm tra các đỉnh còn lại nào có thể được giải thích bằng các peptit có sự phân cắt bị bỏ sót.
-
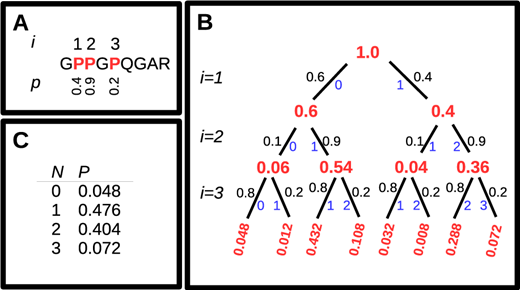
Các sửa đổi sau dịch mã: Với chuỗi peptit thô, cũng cần kết hợp các sửa đổi sau dịch mã phổ biến được quan sát thấy ở collagen. Đây là quá trình hydroxyl hóa proline (P) thành hydroxyproline và khử axit glutamine (Q) để tạo ra axit glutamic (E). Chuỗi chính của collagen được sắp xếp theo thứ tự cao, với mô típ ‘GlyXaaYaa’ lặp lại, trong đó ‘Gly’ là glycine, và Xaa và Yaa có thể là bất kỳ axit amin nào khác. Xác suất hydroxyl hóa proline được biết là có nhiều khả năng xảy ra ở vị trí Yaa trong mô típ lặp lại này, và ít xảy ra hơn ở vị trí Xaa. Các phương pháp tính toán xác suất chính xác của quá trình hydroxyl hóa đối với một proline nhất định trong chuỗi collagen vẫn chưa có sẵn, nhưng các ước tính đơn giản dựa trên vị trí Xaa – Yaa có thể được sử dụng để tính toán xác suất của một số hydroxyl hóa cụ thể, như thể hiện trong phía dưới. Tính xác suất của các mức độ khác nhau của sự kiện hydroxyl hóa trong một peptit. (A) Một peptit có ba proline trong chuỗi của nó. tôi đưa ra chỉ số của mỗi proline và p thể hiện xác suất hydroxyl hóa. Các xác suất này được cung cấp để minh họa cách tiếp cận và không nên được coi là các giá trị điển hình cho chuỗi được trình bày. (B) Cây nhị phân được sử dụng để tính xác suất của tất cả các kết hợp có thể có của hydroxyproline và proline. Tính toán đi từ trên xuống dưới của cây, xem xét lần lượt từng proline. Các số màu đỏ hiển thị xác suất hiện tại khi mỗi proline được xem xét. Các số màu đen cho biết xác suất giảm dần của mỗi nhánh và các số màu xanh lam cho biết số lượng hydroxyl hóa hiện tại của mỗi nhánh. (C) Xác suất thu được cho mỗi mức hydroxyl hóa, đạt được bằng cách tính tổng xác suất ở cuối cây quyết định cho mỗi mức hydroxyl hóa
4.2 Căn chỉnh các peptit lý thuyết với các đỉnh MS
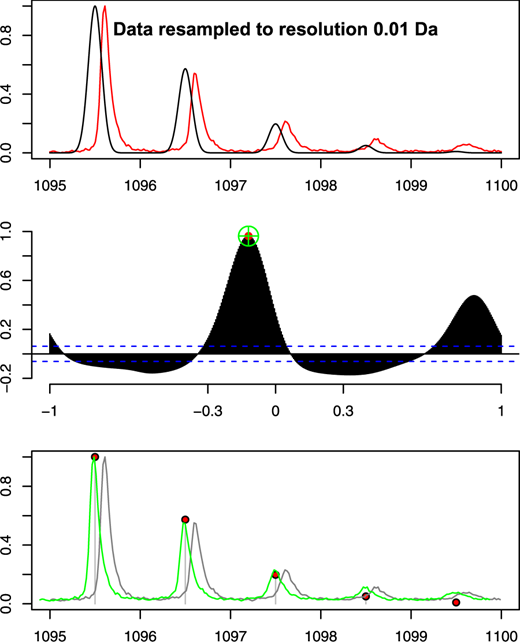
Sau khi tạo ra một tập hợp các đỉnh lý thuyết, bước tiếp theo là căn chỉnh các đỉnh MALDI-ToF quan sát được với khối lượng tính toán. Như đã mô tả trước đó, khối lượng đỉnh MALDI-ToF quan sát được có thể lệch tới 0,5 Da. Chiến lược của ở đây là sử dụng tương quan chéo để tìm ra sự liên kết tốt nhất giữa các khối lượng đồng vị được tính toán của mỗi peptit với vùng cục bộ của phổ MALDI-ToF, kết hợp độ lệch lớn nhất có thể là 0,5 Da. Quá trình này được minh họa trong Hình 3. Dữ liệu MALDI-ToF được lấy mẫu lại thành các khoảng đều đặn 0,01 Da bằng cách sử dụng nội suy tuyến tính. Các giá trị cường độ trong dữ liệu MS được chuẩn hóa thành phạm vi [0,1] . Sự phân bố khối lượng theo lý thuyết của các đồng vị được tạo ra bằng cách thiết lập các giá trị phân bố ion trong dãy giá trị khối lượng - cặp ion có giá trị bằng không với số lượng ion bằng không. Đối với bất kỳ peptit nào, các đỉnh MALDI-ToF không tự biểu hiện thành các đỉnh nhọn đơn lẻ, nhưng có xu hướng hình thành một chuỗi các đỉnh tương đối rộng do hiệu ứng ngẫu nhiên trong quá trình bay của peptit. Đối với một peptit riêng lẻ, các đỉnh của mỗi đồng vị được đặt cách nhau 1 Da. Mối tương quan giữa phân bố lý thuyết và phân bố quan sát được có thể được cải thiện bằng cách áp dụng phép làm trơn Gaussian cho sự phân bố lý thuyết của các đồng vị peptit như thể hiện trong hàng trên cùng của Hình 3.
Kỹ thuật tương quan chéo mang lại hai phần thông tin có thể được sử dụng để đánh giá khả năng một peptit cụ thể đang tạo ra đỉnh trong phổ MALDI-ToF:
- Đầu tiên, điểm tương quan tối đa đưa ra thước đo về mức độ phù hợp của peptit được đề xuất với đỉnh MALDI-ToF.
- Thứ hai, độ trễ cho thấy phổ MALDI-ToF sẽ phải thay đổi bao nhiêu để có được điểm tương quan tối đa.
4.3 Phân loại dựa trên điểm số
Sau khi thu được điểm tương quan cho sự liên kết cục bộ đối với một peptit duy nhất, cần có phương pháp để suy ra phân loại dựa trên các điểm này. Đây không phải là một nhiệm vụ đơn giản, vì chất lượng của sự liên kết phụ thuộc vào chất lượng của các đỉnh mà lần lượt phụ thuộc vào toàn bộ lịch sử của collagen đang được xem xét — từ khi hình thành trong cơ thể sống, cách nó được bảo quản (thường trong nhiều thế kỷ ) đến cách chuẩn bị mẫu để phân tích. Kinh nghiệm của các chuyên gia cho thấy rằng không khả thi khi xác định một ngưỡng tổng thể duy nhất trên điểm tương quan mà dưới đó, kết quả phù hợp căn chỉnh bị từ chối, vì bất kỳ ngưỡng nào đều có xu hướng tạo ra kết quả dương tính giả và âm tính giả do mức độ biến thiên cao của các đỉnh trong MALDI-ToF. Phân tích của các chuyên gia chỉ tốt khi có dữ liệu thô, và nếu việc bảo quản collagen kém thì không phải lúc nào cũng có thể thực hiện được.
Một cách tiếp cận khác mạnh mẽ hơn là đếm và kết hợp các lần ghép cặp so sánh có điểm tương quan trên một phạm vi ngưỡng và phát triển hệ thống tính điểm dựa trên phân tích phạm vi này. Vì điểm tương quan luôn nằm trong phạm vi [0,1] , nên khả thi khi sử dụng ngưỡng này làm trọng số cho số lần truy cập ở mức ngưỡng đó. Trong mô tả bên dưới, 'lần truy cập - hit' là sự liên kết có điểm tương quan cao hơn một ngưỡng cụ thể.
Chiến lược phân loại
Chiến lược phân loại của các chuyên gia kết hợp bằng chứng cho mỗi bản sao của một mẫu khi được căn chỉnh với một tập hợp các peptit đánh dấu cho từng loài mục tiêu. Phương pháp tính điểm cho mỗi loài ứng viên bằng cách xem xét số lượng liên kết peptit trên ngưỡng tương quan chạy từ 0 đến 1 với gia số 0,05. Bắt đầu bằng cách xác định các biến sau:
- r ∈ R là một trong ba bản sao;
- p ∈ P là tập hợp các peptit cho mỗi đơn vị phân loại i;
- Ngưỡng tương quan Ct;
- i ∈ I là tập hợp các đơn vị phân loại mà từ đó việc xác định được rút ra;
- H(i, t) là số lần truy cập cho mỗi kết hợp peptit / bản sao đối với đơn vị phân loại i và ngưỡng tương quan Ct.
Đối với mỗi lần lặp lại r, quang phổ sắp xếp theo từng peptit p cho mỗi đơn vị phân loại i. Căn chỉnh được coi là 'lần truy cập - hit' và đạt điểm 1 nếu điểm tương quan c(r, p) lớn hơn ngưỡng Ct:
Phương trình tổng có dạng sau:
Do đó, S cao khi một đơn vị phân loại cụ thể có nhiều lần truy cập hơn các đơn vị phân loại khác đang được xem xét, nhưng tương đối thấp nếu mẫu không rõ ràng. Bằng cách này, bằng chứng để xác định một mẫu được tích lũy bằng cách sử dụng một loạt các ngưỡng tương quan. Ưu điểm của thước đo S là: nó cho điểm tin cậy để kiểm tra tất cả các đơn vị phân loại đang được xem xét; cao đối với mẫu tốt và thấp đối với mẫu xấu; nó chỉ cao khi có thể có nhiều hơn một cách diễn giải; và sẽ thấp nếu mẫu từ một loài không được xem xét. Quá trình này cho phép thực hiện phân tích hoàn toàn tự động sau khi có mẫu.
5. Kết luận
Bài viết này mình đã tìm hiểu được cơ chế nghiên cứu tự động và của các chuyên gia để xác định một vật mẫu có nguồn gốc từ loài động vật nào thông qua việc kiểm tra quang phổ MALDI-ToF. Ở bài viết sau, chúng ta sẽ cùng cài đặt tool mà bài báo giới thiệu để thử phân loại nhé!!!
Cảm ơn mọi người đã dành thời gian đọc bài viết (bow)!
Link github tool ở đây: bacollite - tool.
Tài liệu tham khảo
Bài báo khoa học: How to get your goat: automated identification of species from MALDI-ToF spectra.
All rights reserved
